Traduzione di Natale
Marzari
Traduzione di Natale
Marzari
Home,
Ricerca,
Indice alfabetico,
Collegamenti,
Patologia,
Molecole,
Sindromi,
Muscoli,
Giunzioni neuromuscolari,
Nervi,
Spinale,
Atassia,
Anticorpi e Biopsia,
Informazioni per i
pazienti
|
 |
Traduzione del 6 settembre 2010
DISTURBI DELL'AUTONOMO
SINDROMI DA MALATTIE DELL'AUTONOMO
- Malattie sistemiche
- Disturbo immunitario
- Degenerazioni sistemiche
- Disturbi ereditari dell'autonomo
- Anestesia congenita di Biemond

- Dolore (sottomandibolare, oculare e rettale) con arrossamento
- Altre disautonomie congenite 1
- Sudorazione ridotta
- Sincope
- Vomito
- Salivazione
ridotta
- Difficoltà ad alimentarsi durante l'infanzia
- Ritardo nella crescita
- Lacrimazione ridotta
-
Anomalie del controllo della temperatura
- Farmaci e tossine
- Polineuropatia
- Disfunzione cellulare: FBracciaacologica
- Riducono l'attività dell'ortosimpatico
- Neuroni dell'ortosimpatico: Guanetidina; Betanidina
- Bloccano gli adrenocettori α: Fenossibenzamina; Prazosina
- Bloccano gli adrenocettori β: Propranololo; Timololo
- Aumentano l'attività dell'ortosimpatico
- Rilascia noradrenalina: Tiramina
- Inibitori della monoamina ossidasi
- Stimolanti degli adrenocettori β: Isoprenalina
- Decrementano l'attività del parasimpatico
- Aumentano l'attività del parasimpatico
- Acetilcolinomimetici
- Carbacolo; Betanicolo; Pilocarpina
- Molluschi: Muscimolo e Muscarina
- Anticolinesterasici
- Iperattività dell'autonomo
- Primario Disautonomia: Spesso monofasico + remissione
- Acuta
- Insufficienza cronica dell'autonomo
- Congenita: Carenza del fattore di crescita nervosa
- Disfunzione localizzata dell'autonomo
SINDROMI DELL'AUTONOMO: Disturbi sistemici
Pandisautonomia: Acuta 16
- Insorgenza
- Età: Adulti o bambini
- Segni
- Femmine > maschi 2:1
- Progressione 1 ÷ 8 settimane
- "Prodromi virali" nel 59%: Specialmente URI o Sindromi simil-influenzali
- Persone precedentemente sane
- Caratteristiche anatomiche
- Distribuzione della disfunzione dell'autonomo: Diffuse; Troncale; Scalpo
- Sistemi autonomi coinvolti: Ortosimpatico e parasimpatico nel 80%
- Caratteristiche cliniche
- Laboratorio
- CSF: Proteine elevate nel 60%; Non cellule
- Anticorpi: IgG vs subunità α3 dei recettori dell'acetilcolina 4
(30%)
- Titolo anticorpale più alto (≥ 1 nmol/L) correlato con
- Caratteristiche dell'autonomo: Presenti e più gravi
- Neoplasmia
- Adenocarcinoma (43%): Mammella > Prostata, polmoni e GI
- linfoide (17%)
- Altri disturbi con anticorpi anti-α3-AChR: Specialmente con titolo intermedio
- Titolo basso (0.03-0.09 nmol/L): Non specifico
- Associazione comune con anticorpi
- GAD65
- AChR muscolari
- Canale cationico controllato dal voltaggio: N-tipo Ca++; K+; P/Q
tipo Ca++
- Nigrostriatale
- Hu
- CRMP-5
- Vedi anche: Disturbi da AChR
- Patologia nervosa: Occasionali infiammazioni perivascolari
- Malattie associate
- Neoplasmia: 20%; Specialmente con
alto titolo IgG vs AChR neuronali
- Carcinoma polmonare a piccole cellule: Le cellule possono contenere AChR neuronali
- Vescica
- Rettale
- Diabete: 10% delle neuropatie autonome diabetiche
- Diagnosi differenziale
- Recupero
- Parziale: L'esito più comune
- Lento nel corso di mesi o anni;
- Correlato con: Riduzione del titolo anticorpale
- Diminuito bloccaggio della funzione degli AChR
- Nessuno in alcuni pazienti
- Occasionale recupero completo
- Trattamento
- Per ciascuna neoplasmia associata
- FBracciaacologica: ? IV Ig; Specialmente la 1° di 8 settimane della malattia o
della progressione della disabilità
- Modello animale: Conigli immunizzati vs le
subunità ricombinanti α3 nAChR 15
- Caratteristiche cliniche: Ipomotilità gastrointestinale; Pupille dilatate con insufficiente risposta allo stimolo luminoso; Vescica distesa
- Gravità parallele ai livelli serici degli autoanticorpi nAChR ganglionici
- Difetto post-sinaptico
- Insufficienza della neurotrasmissione attraverso i gangli ortosimpatici addominali
- Ritenzione della viabilità neuronale
Sindromi gastrointestinali
- Megacolon congenito di Hirschsprung
- Caratteristiche generali
- Patofisiologia
- Insufficiente migrazione nel tempi specifici delle creste neurali derivati
dalle cellule gangliali dentro il tratto GI
- Percorsi genetici coinvolti
- Recettore per la tirosina chinasi: Recettore RET; GDNF (ligando del recettore RET)
- Recettore endotelina tipo B: EDNR; Endotelina 3 (ligando EDNR)
- Trascrizione mediata da SOX-10
- Insorgenza: Congenita
- Modelli di ereditarietà: Malattia non sindromica può essere oligogenica
- Aganglionosi solamente nel colon sigmoide: Spesso sporadica; 60%
÷ 85% dei casi
- Aganglionosi per tutta la lunghezza dell'intestino: Più probabile ad incorrenza familiare
- Epidemiologia
- Incidenza: 1 su 5.000 neonati
- Predominanza maschile: 4 a 1
- Ricorrenza in fratelli e sorelle: Solitamente 3%
÷ 4%; 200x più alta che nella popolazione generale
- Caratteristiche cliniche
- Costipazione
- Vomito
- Dilatazione addominale
- Ostruzione intestinale
- Associazione con malformazioni congenite (30%)
- Formazione di pigmenti craniali
- Componenti sensori dei percorsi acustici
- Patologia
- Assenza dei gangli parasimpatici enterici lungo tratti di variabile lunghezza dell'intestino
- Incompleta migrazione delle cellule gangliali neuroenteriche dalle creste neurali
all'intestino
- Acetilcolinesterasi: Aumentata nei plessi mienterico e sottomucosale dei
segmenti affetti
- Restringimento del segmento distale dell' intestino
- Diagnosi: Assenza o riduzione delle cellule gangliali nei segmenti affetti
nella
biopsia rettale
l
Oncogene RET (HSCR1)
; Cromosoma 10q11.2;
Dominante o sporadica
- Frequenza: 50% dei casi familiari; 15% ÷ 35% dei casi sporadici
- Mutazioni del gene
- Famiglie multigenerazionali: Le mutazioni avvengono fra i geni
- Casi sporadici: Mutazioni nella regione codificante dominio extracellulare
- Meccanismi genetici 10
- Perdita di funzione
- Associazione con altre mutazioni genetiche
- Malattia più grave, segmenti più lunghi con addizionali mutazione Neurturina
- Altre associazioni al locus genetico (suscettibilità)
- Localizzazioni
- HSCR6
; Cromosoma 3p21
- HSCR7
; Cromosoma 19q12
- Trasmissione: Materna > paterna
- Amplificazione degli effetti: Moltiplicativa fra i loci
- Penetranza: 50% (Femmine), 72% (Maschi)
- Clinica
l
Endotelina, recettore tipo B (EDNRB; HSCR2)
; Cromosoma 13q22;
Recessiva
- Genetiche
- Penetranza: 30% ÷ 85%
- Allelica con: La sindrome di Waardenburg-Shah (WS4)
- Clinica
- GI (Hirschprung's): Malattia intestinale a segmenti corti nel 95%
- Sordità
- Anomalie pigmentose
l
GDNF (HSCR3)
; Cromosoma 5p13.1-p12;
Dominante o recessiva
l
Endotelina 3 (EDN3; HSCR4))
; Cromosoma
20q13.2-q13.3; Recessiva o dominante con penetranza ridotta
- Genetica: Allelica con: La sindrome di Waardenburg-Shah (WS4)
- Ipoventilazione centrale congenita con la malattia di Hirschsprung
- Insufficiente ventilazione autonoma nel sonno
- GI: Megacolon (Spesso aganglionosi per l'intera lunghezza); Ridotta motilità esofagea
- Faccia: Angolazione degli occhi antimongoloide; Bocca triangolare
- Orecchie situate più in basso; Naso sottile
- Controllo del ritmo cardiaco: Ridotto
- Diagnosi differenziale
- Depressione della risposta ventilatoria alla ipossia e ipercapnia
- Apnea nel sonno familiare letale
l
Neurturin
Cromosoma 19p13.3; Sporadica
- ? Alcuni casi hanno ereditarietà digenica con RET
l
Endotelina, enzima di conversione (ECE1)
; Cromosoma 1p36.1;
Dominante o recessiva
- Malattia di Hirschsprung: Lesioni da lacune
- Difetti cardiaci
- Anormalità craniofacciali + Altre caratteristiche dismorfiche
- Disfunzione dell'autonomo
l
SOX10
; Cromosoma 22q13;
Dominante o sporadica (Recessiva)
- Ipomielinizzazione in CNS e PNS
- Ipopigmentazione
- Sordità
- Aganglionosi enterica
- Allelica con
l
SMADIP1 (omeobox dita a zinco 1B; ZFHX1B; ZEB2)
Cromosoma 2q22; Dominante o recessiva
- Nosologia
- Mowat-Wilson sindrome
: Solitamente dominante
- Simile alla sindrome ad ereditarietà recessiva: Goldberg-Shprintzen
- Genetica: Mutazioni
- Frameshift o nonsenso
- Sindrome da delezione: Pazienti occasionali
- Proteina SMADIP1 (ZFHX1B)
- Proteine omeodominio/dita a zinco 2-handed (omeobox dita a zinco 1B)
- SMADIP1 interagisce mediata dal recettore, attivata per l'intera lunghezza
della SMAD
- Clinica
- Può manifestarsi da sola (Non sindromica) o con altra sistemica
- Neonatale : Ipotonia; Megacolon di Hirschsprung
- Fenotipo facciale: Ponte nasale ampio (Ipertelorismo); Bocca aperta;
mascella triangolare
- EENT: Occhi grandi e incassati (Blu); Lobi delle orecchie grandi e
situati in alto
- CNS
- Ritardo mentale: Parlata minima; Ritardo motorio
- Epilessia
- Problemi di apprendimento
- Patologia del CNS: Microcefalia; Agenesi del corpo calloso
- Oculare: Coloboma dell'iride; Ptosi; Iridi bicolore
- Malattia cardiaca: congenita
l
KIAA1279
 ; Cromosoma 10q22.1;
Recessiva
; Cromosoma 10q22.1;
Recessiva
- Nosologia
-
Sindrome di Goldberg-Shprintzen (GOSHS)
con
polimicrogiria 22
- Simile alla sindrome ad ereditarietà dominante: Mowat-Wilson
- Epidemiologia: Famiglia marocchina
- Genetica
- Mutazioni di stop omozigotiche: Arg90X; Glu84X
- KIAA1279
- Famiglia delle ripetizioni tetratrico peptidiche (TPR)
- Espressione ubiquitaria
- Altri disturbi della famiglia TPR
- Clinica
l
Associata alla sindrome di Down
- ? Correlata al modificatore genetico di Hirschsprung (HSCR5)
nel Cromosoma 21q22
l
Modificatore genetico di Hirschsprung 8 (HSCR8)
Cromosoma 9q31
l
Modificatore genetico di Hirschsprung 9 (HSCR9)
Cromosoma 4q31-q32
l
Altre sindromi di Hirschsprung
- Polidattilia, agenesi renale, sordità
- Unghie ipoplastiche o deformità delle dita
- Neoplasia endocrina multipla 2A
; 2B
e carcinoma tiroideo midollare
- Difetti cardiaci, anomalie laringee, polidattilia preassiale
- Brachidattilia di tipo D
- Pitt-Hopkins
- Acalasia
- Definizione
- Assenza of peristalsi nell'esofago
- Insufficiente rilassamento dello sfintere esofageo inferiore
- Eziologia
- Solitamente 1° disturbo
- Occasionalmente 2° a Chagas
- Patologia
- Perdita di cellule gangliali e dei nervi mienterici
- Ridotto numero neuroni plessomienterici contenenti peptidi intestinali
vasoattivi
- Neoplasia endocrina multipla 2b (o 3)
l
Oncogene RET
; Cromosoma 10q11.2;
Dominante
- Ganglioneuromi: Lingua e labbra
- Dismotilità GI: esofago, megacolon
- Neoplasmie: Tiroide midollare; Feocromocitoma
- Miopatia viscerale familiare con oftalmoplegia esterna
- Occhi: Ptosi e oftalmoplegia
- GI: Pseudoostruzione intestinale; Asciti
- Progressione: Morte spesso < 30 anni
- Patologia:
- Perdita di muscolatura liscia in stomaco e intestini
- Neuropatia periferica
- Neuropatia viscerale famigliare
l
Recessiva
- Insorgenza: 1a ÷ 2a decade
- Clinica
- Pseudoostruzione intestinale
- Altre: Alcuni pazienti con caratteristiche del PNS o CNS
- Atassia
- Pupille: Scarsamente reattive
- Riflessi tendinei ridotti
- Perdita sensoria: Vibrazioni e senso di posizione
- Neuropatia viscerale famigliare
l
Dominante
- Clinica
- GI
- Pseudoostruzione intestinale
- Acalasia esofagea
- Neurologico exan: Spesso normale
- Patologia: Plessi mienterici
- Neuroni: Morfologia anormale; Numero aumentato
- Nervi del tronco: Iperplastici
- Ipoplasia capelli cartilagine (CHH)
l
Endoribomucleasi
mitocondriali
processante RNA, componenti RNA (RMRP)
; Cromosoma 9p21-p12;
Recessive
- Allelica con
- Displasia metafisiaria senza ipotricosi
- Displasia anaussetica
- RMRP
- Gene RMRP: Non traslato, codifica RNA, non proteine
- Ribonucleoproteina: Contiene proteine e componenti RNA
- RNA codificato da geni a singola copia nel nucleo: Importato dentro i mitocondri
- Attività dell'enzima
- Endoribonucleasi
- Stacca l'RNA mitocondriale complementare dall'ansa di collocazione della
catena leggera
- Epidemiologia: Amish del Vecchio Ordine (US); Finlandia
- Clinica
- Scheletrico
- Bassa statura: Sproporzionato
- Iperestensibilità delle articolazioni: Mani, polsi, e piedi
- Displasia metafisiaria: Può essere la sola manifestazione
- Capelli: Ipoplastici; Fini, sparsi e colorati leggermente
- Intestino: Displasia neuronale; Megacolon; Malassorbimento
- Malignità: Aumentata rischio di linfoma e neoplasmia cutanea
- Laboratorio
- Immunità difettosa: Suscettibilità alla varicella; Linfopenia;
Neutropenia
- Anemia: Ipoplasia macrocitica
- Disturbi gastrointestinali acquisiti
- Malattia di Chagas (Tripanosomiasi
cruzii)
- Sudorazione gustatoria (sindrome di Frey)
- Paraneoplastica
- Post-chirurgica
- Simpatectomia regionale
- Dumping sindrome: Post-vagotomia e drenaggio gastrico
- Trapianto cardiaco
Ritmo cardiaco: Controllo parasimpatico
- Tachicardia
- Bradicardia
- A riposo: Carenza di dopamina idrossilasi β
- Parasimpatico rispBracciaiato
- Risposta allo stimolo
- Ipersensibilità del seno carotideo
- Sincope deglutiva e vasovagale
- Espirazione (Vagale; )
- Cardiomiopatia: Emery-Dreifuss
Colinergica
Ipotensione posturale
(Pressione sanguigna:
controllo noradrenergico dell'ortosimpatico)
- Causa
- Esacerbazione
- Rapidi cambiamenti di posizione
- Al mattino
- Grossi pasti
- Prolungata positura supina
- Ambiente caldo
- Aumento della pressione intratoracica: tosse; minzione; defecazione; esercizio
- Farmaci vasoattivi
- Trattamento
- Evitare i fattori esacerbanti
- Il coprirsi
- Ridurre la perdita di sale: Mineralcorticoidi
- Fluoroidrocortisone 0,1 mg qd a qid
- Vasocostrizione
- Resistenza dei vasi
- Midodrina 10 mg bid o tid
- Fenilefrina
- Fenilpropanolamina 25 mg tid
- Capacità dei vasi: Diidroergotamina
- Indiretti: Efedrina; inibitori del MAO; Ioimbina
- Prevenire la vasodilatazione
- Inibitori della sintetasi delle prostaglandine: Indometacina; Flurbiprofene
- Blocco dei recettori della dopamina: Metoclopramide; Domperidone
- Blocco dell'adrenocettore 2 β: Propranololo
- Prevenire l'ipotensione postprandiale
- Evitare il riempimento gastrico: Pasti più leggeri e più frequenti
- Blocco del recettore dell'adenosina: Caffeina 250 mg
- Caffè o te forti prima di alzarsi dal letto ed ai pasti
- Bere acqua 9
- Agisce rapidamente (Minuti)
- Prima dei pasti: 120 ÷ 480 ml in 5 minuti
- Assunzione totale giornaliera: 2 ÷ 3 litri
- Media attraverso l'attivazione dell'ortosimpatico: Aumenta la norepinefrina plasmatica
- Inibitori del rilascio peptidico: Analoghi alla somatostatina; Octreotide
- Altri fBracciaaci: Ibuprofene 400 mg; Fenilpropanolamina 25 mg; Ergotamina intranasale: 1 o 2
sbuffi
- Aumentano la gittata cardiaca: Pindololo; Xamoterolo
- Aumenta la massa dei globuli rossi: Eritropoietina
- Riduce poliuria notturna: Desmopressina
- Riduce la diminuzione della pressione diastolica: Piridostigmina (60 mg)24
- Evitare l'ipertensione supina:
- Ridurre l'eccesso di fluoroidrocortisone
- Non vasocostrittori dopo le 18.00
- Dormire con il capo rialzato dal letto
- Trattamento: Enalaprile (inibitore dell'ACE) con la dose massimale al momento di andare a letto
Altri disturbi della pressione sanguigna
- Geni del
canale sodio epiteliali SCNN1A
; SCNN1B
; SCN1G
- Ipotensione: Pseudoipoaldosteronismo, Tipo 1
- Ipertensione: Sindrome di Liddle
Sudorazione: Ortosimpatico; Controllo colinergico muscarinico
- Iperidrosi
- Meccanismi
- Aquaporina 5 (AQP5)
nella membrana plasmatica delle ghiandole sudoripare necessaria per secrezione del sudore
- Disturbi generali
- Compensatoria per perdita della sudorazione in altre parti
- Può essere il sintomo iniziale con progressione ad anidrosi
- Tossici
- Altre acquisite
- Sindrome POEMS
- Neuromiotonia
- Holmes-Adie
- Sindrome arlecchino
- Sudorazione gustatoria (sindrome di Frey)
- Aberrazioni dell'ortosimpatico, assoni colinergici afferenti alle ghiandole sudoripare facciali
- Iperidrosi segmentale
- Cause: Siringomielia; Disturbi vascolari; Neoplasmie
- Trattamenti possibili: Tossine botuliniche; Mexiletina
- Hirayama
- Ereditaria
- Anidrosi
- Dovuta a danneggiamento di
- Assoni colinergici dei nervi periferici non mielinizzati, postgangliare,
- Assoni pregangliari efferenti dell'ortosimpatico
- Ghiandole sudoripare
- Diagnosi differenziale
- Displasia ectodermica anidrotica (ED1)
l
Ectodisplasina-A (EDA)
); Cromosoma
Xq12.2-q13.1 Recessiva
- Proteina: Piccola, 135 AA, transmembrana
- Clinica
- Capelli: Sparsa
- Dentizione: Anormale o mancante
- Anidrosi: Dovuta a mancanza delle ghiandole sudoripare
- Ipertermia: Pericolosa per la vita o per danni al cervello o cervello
- Femmine portatrici: Dentizione anormale
- Displasia ectodermica ipoidrotica con immunodeficienza
l
IKK-γ (IKBKG; NEMO)
; Cromosoma ; Recessiva
- Genetica
- La mutazione produce la parziale perdita della funzione di IKK-γ
- Allelica con l'incontinenza dei pigmenti
: Completa perdita della
funzione di IKK-γ
- Proteina IKK-γ
- Necessaria per l'attivazione del fattore nucleare κ fattore di trascrizione B
- Ruolo nelle funzioni delle cellule T e B
- Clinica
- Insorgenza: < 2 anni; Infezioni
- Ipoidrosi
- Dentizione anormale
- Osteopetrosi
- Immunodeficienza
- Infezioni frequenti
- Prognosi: Spesso morte precoce
- Displasia ectodermica anidrotica
l
Dominio morte associato a
EDAR (EDARADD)
; Cromosoma 1q42.2-q43;
Recessive
- Proteina EDARADD
- Espresso nelle cellule epiteliali durante la formazione dei follicoli piliferi e
dei denti
- EDARADD auto-associata
- Proteina del dominio morte
- Interagisce con il dominio morte di EDAR
- Collega il recettore ai percorsi dei segnali efferenti
- Clinica: Simile a AED
collegata a X
- Topo: Fenotipo 'Crespo'
- Displasia ectodermica anidrotica
l
Ectodisplasina 1, recettore anidrotico (EDAR)
; Cromosoma 2q11-q13;
Recessiva
o dominante
- Proteina EDAR
- Similarità strutturale a TNF
- Attivatore di NF-kappa-B (vedi 164011) attraverso la proteina NEMO
- Clinica
- Simile a AED collegata a X
- Morfogenesi anormale dei denti, dei capelli e delle ghiandole sudoripare esocrine
- Displasia ectodermica anidrotica + immunodeficienza delle cellule T
l
Gene della catena leggera kappa del fattore nucleare potenziatore dell'inibitore
α delle cellule B;
(NFKBIA)
; Cromosoma 14q13;
Dominante
- Mutazione
- Proteina NFKBIA
- Inibisce il complesso NFKB
- Inattiva NF-kappa-B by intrappolandolo nel citoplasma
- Clinica
- Multiple infezioni
- Denti conici
- Cute: Secca; Rugosa
- Laboratorio
- Biopsia cutanea: Assenza delle ghiandole sudoripare
- Displasia ectodermica anidrotica: Altre
l
Con sordità
l
Con ritardo
- Displasia ectodermica idrotica (ED2)
l
Proteina del Gap di giunzione β 6 (GJB6,
Connessina-30, CX30)
; Cromosoma 13q12;
Dominante
Urinario
- Nicturia: Ipotensione posturale
- Rilocazione di liquidi dalla periferia
- Trattamento: Desmopressina
- Nicturia: Disautonomia colinergica
- Atonia della vescica
- Comune nella sindrome di Shy-Drager (Atrofia multisistemica):
Lesione nel nucleo sacrale di Onuf'
- Trattamento
- Minzione programmata con tecnica a doppio svuotamento
- Pulire, con intermittente auto-cateterizzazione
- Prevenire le infezioni
- HSAN IIB
- Sindrome di Parkinson
Sessuale disfunzione
- Erezione: Dipendente dal supporto del parasimpatico e dall'ossido nitrico
- Trattamento
- Iniezioni intracavernose di
- Prostaglandina E, o papaverina ± fentolamina
- Crema topica con nitroglicerina
- Fisico: Intervento chirurgico
- Eiaculazione: Dipendente dal sistema ortosimpatico
Oculare
- Sindrome di Horner

Di: P Bailey
|
- Caratteristiche cliniche
- Ptosi
- Parziale: Non copre mai il campo visivo
- Può interessare sia le palpebre superioni (muscoli di Müller) che quelle
inferiori (sollevate)
- Pupille
- Miosi
- Più evidente all'oscuro
- Mai grave
- Dilatazione: Lenta; Scarsa nell'oscurità
- Diagnosi differenziale di pupille molto piccole
- Gocce miotiche
- Disturbo del CNS
- Adie
- Pupille di Argyll Robertson
- Ipoidrosi
- Lesioni pregangliari prossimali alla biforcazione carotidea
- Distribuzione: Faccia e capo
- Iperemia: Congiuntiva e palpebre; Con lesioni acute
- Vasodilatazione facciale: Variabile
- Enoftalmo: Apparenza ma raramente vero
- Congenita di Horner
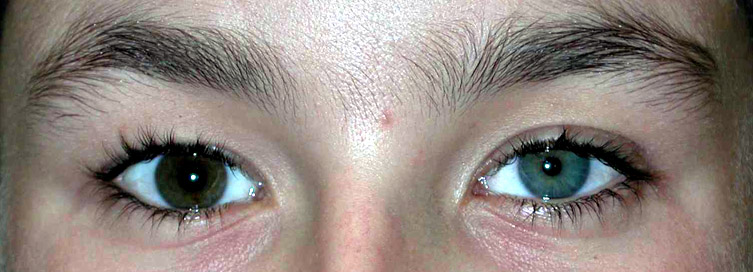
- Eterocromia: Iride blu dal lato della lesione
- Associazioni: Polineuropatia ereditaria
- Amiloide: Bilaterale (50%)
- HSAN-I: Occasionale; unilaterale
o bilaterale
- Lesioni
- Percorsi omolaterali dell'ortosimpatico
- CNS
- Sindrome midollare laterale
- Occasionale midollo spinale cervicale
- Pregangliare
- Localizzazione: Mediastino; Apici polmonari; Spazio sopraclavicolare
- Lesioni
- Spesso neoplasmia
- Solitamente con coinvolgimento dei plesso brachiale
- Lesione iniziale ®
iperfunzione con miridiasi e iperidrosi
- Postgangliari: Ptosi, Miosi, Dolore e sudorazione omolaterale preservata
- Localizzazione: Plesso carotideo lungo l'arteria carotidea interna
- Intracraniche (seno cavernoso) o trombosi interna della carotidea
- Sindrome paratrigeminale: Dolore nella faccia e malattia di Horner
- Esaminazione fBracciaacologica: Comparare le risposte nei 2 occhi
- Nessuna dilatazione pupillare da cocaina
- Postgangliari: Nessuna dilatazione con idrossianfetamina all'1%
- Sindrome di Holmes-Adie
- Ereditaria
- Acquisita
- Più comune nelle giovani donne
- Bambini: Spesso con storia di varicella
- Cause specifiche
- Patologia
- Parasimpatico, post-gangliare, denervazione
- Lesioni: Gangli ciliari; Nervi ciliari posteriori
- Caratteristiche cliniche
- Pupille: "Toniche"
- Dilatate
- Miotonica
- Luce: Lentamente o non reattive
- Sforzo di avvicinamento
- Rifissazione a distanza: Dilatazione lenta
- Patologia: Cellule nei gangli
ciliari parasimpatici ridotte
- Settori di residua reazione alla luce quando illuminate dalla lampada a
fessura
- Ariflessia: ? Dovuta a patologia nei gangli della radice dorsale o colonna
posteriore del midollo spinale
- Altre caratteristiche associate
- Sindrome di Ross 13
- Insorgenza: 1° ÷ 3a decade
- Caratteristiche cliniche
- Anidrosi
- Segmentale o generalizzata
- Residua sudorazione: Più comune nel tronco; Iperidrosi in
queste aree
- Decorso: Perdita progressiva; Le aree possono essere iperidrotiche prima della perdita della sudorazione
- Intolleranza al calore
- Iporiflessia: Specialmente alle caviglie
- Pupille toniche
- Reazione lenta alla luce
- Normale costrizione vicino all'obiettivo
- Altre disfunzione dell'autonomo: Cardiaca in alcuni pazienti
- Sensorio
- Alterazioni subcliniche
- Ridotte sensazioni di temperatura, dolore e tattile in alcuni pazienti
- Elettrofisiologia
- Riflesso H: Assenti
- Conduzione nervosa: Normale
- Risposte cutanee ortosimpatiche: Assenti
- Patologia
- Denervazione post-gangliare del parasimpatico : Sudomotoria
- Assoni sensori epidermici
- Perdita di fibre mielinizzate e non mielinizzate
- Ridotta innervazione delle ghiandole sudoripare, dei vasi sanguigni e
dei muscoli erettori piliferi
Assenza di lacrimazione
Disturbi respiratori, dell'autonomo
- Sindrome da ipoventilazione centrale congenita (CCHS)
- Generale
- Spesso associata con altri disturbi dell'autonomo
- Hirschsprung: 30%
- Nosologia: Apnea notturna
- Criteri clinici
- Ipoventilazione, ipossiemia e ipercapnia durante il sonno tranquillo
- Nessuna anormalità cardiaca, polmonare, neuromuscolare, EEG o della MRI cerebrale
- Associazioni genetiche
l
Endotelina 3
; Cromosoma
20q13.2-q13.3; Recessiva o dominante con penetranza ridotta
l
Oncogene RET
; Cromosoma 10q11.2;
Dominante o sporadica
- Vedi anche: Hirschsprung
- Malformazione del sistema nervoso enterico
l
GDNF
; Cromosoma 5p13.1-p12;
Dominante (HSCR1) o recessiva
l
BDNF
; Cromosoma 11p13;
Dominante
- CHS isolata
- Padre con ipotensione posturale e sincope vasovagale
l
Omeobox mesodermici appaiati 2B (PHOX2B; PMX2B)
; Cromosoma 4p12;
Dominante
- La più comune mutazione genetica nelle sindromi da ipoventilazione congenita: 60%
÷ 80%
- Nuove mutazioni in 1° generazione
- Possono essere associate con Hirschsprung e neoplasmie
- Mutazioni 21
- Alcuni pazienti con mutazioni di espansione polialaniniche
- Il tipo di mutazione più comune
- Solitamente duplicazione in-frame
- Aggiunge 5 ÷ 13 alanine al tratto normale
delle 20 polialanine residue
- Mosaicismo somatico
- Mutazione della stessa dimensione in differenti generazioni
- Malattia a tarda insorgenza: Dimensione delle inserzioni 5 alanine
- Mutazioni frameshift o missenso: Molte presenti con
- Malattia di Hirschsprung
- Tumori del sistema nervoso ortosimpatico
- Tipi: Neuroblastoma, Ganglioneuroblastoma, Ganglioneuroma
- Localizzazione: Tumori spesso multifocali
- Meccanismi della malattia
- Mutazioni alaniniche con "aumento della funzione": Aggregazione citoplasmatica di proteine normali e mutate
- Le mutazioni con aploinsufficienza: Cause pupille dilatate e atrofia dei
gangli ciliari
l
Achaete-scute umana omologa 1 (HASH-1; ASCL1)
; Cromosoma 12q22-q23;
Dominante, penetranza incompleta
- Mutazioni: Alcuni consistono in perdita di alanine
nel tratto polialaninico
18
- Normale: 13 alanine
- Mutazioni: 5 o 8 alanine
- Espressione della proteina HASH 1
- CNS
- PNS
- Sviluppo: Sistema nervoso enterico, dall'esofago al retto; Midollo adrenale
- Neuroblastoma
- Funzione della HASH 1
- Sviluppo dei neuroni olfattivi e della maggior parte dei neuroni dell'autonomo
periferici
- Formazione dei circuiti neuronali entro il CNS
- La mutazione produce un insufficiente sviluppo neuronale noradrenergico
- Interagisce con la proteina E2 2
- Sindromi cliniche
- Ipoventilazione centrale
- Sindrome di Haddad
- Ritardo mentale con intermittente iperventilazione (sindrome di Pitt-Hopkins)
26
l
TCF-4
; Cromosoma 18q21.1;
Dominante o sporadica
- Genetica: Mutazioni
- Eterozigotica
- Tipi: Microdelezione; Missenso; Stop; Sito di splice
- Proteina TCF-4
- Proteina E: Fattore di trascrizione basico elica-ansa-elica classe I
- Localizzazione subcellulare: Nucleare
- Espresso nel cervello fetale e adulto
- Meccanismo della malattia
- Aploinsufficienza
- ? Danneggiato lo sviluppo noradrenergico e altro neuronale
- Complessi con ASCL1
- Clinica
- Insorgenza: Infanzia
- CNS
- Ritardo psicomotorio: Grave
- Epilessia
- Iperventilazione: Episodi quotidianamente; Diurni
- Ipotonia
- Morfologia
- Ritardo nella crescita: Lieve
- Microcefalia: Postnatale
- Faccia
- Naso: Grande; Ponte alto; Narici arrossate
- Bocca: Ampia; Labbra carnose; Palato ampio; Denti ampiamente spaziati;
arco di cupido a forma di M
- Orecchie: Elici displastiche e spesse
- Occhi
- Enoftalmia
- Strabismo
- Sopraciglia: Assottigliate nella porzione mediana
- GI: Alcuni pazienti
- ? Malignità: Linfoma nei pazienti più anziani
- CNS patologia
- Cerebello: Ipoplasia di
emisferi e verme
- Corpo calloso: Ipoplasia
- Ippocampo: Piccolo
- Nuclei caudati: Rigonfi
- Sindromi di Joubert
Atrofia multisistemica (Shy-Drager)
l
Sporadica o autosomica dominante
- Nosologia: Altri disturbi simili o sovrapposti
- Degenerazione nigrostriatale
- Atrofia olivopontocerebellare sporadica ad insorgenza in età adulta (OPCA)
- Sindrome Shy-Drager
- Caratteristiche cliniche
- Insorgenza: Adulti; Parkinsonismo precoce e disfunzioni della vescica
- Autonomo
- Ipotensione ortostatica
- Incontinenza della vescica
- Anidrosi; Ptosi
- Disfunzione intestinale
- Extrapiramidale
- Parkinsonismo (87%): Specialmente rigidità, instabilità posturale e
bradicinesia
- Risposta distonica alla L-DOPA
- Segni dei motoneuroni superiori (50%): Spasticità;
Riflessi tendinei vivaci
- Bulbare
- Parlata sillabata
- Stridore: Paralisi degli adduttori laringei (anche vedi
atassia recessiva)
- Disturbi cerebellari (Specialmente variante OPCA): 50%
- Miocloni: Sensibili allo stimolo; Arti e faccia
- Sistema nervoso periferico
- Segni dei neuroni motori inferiori (20%)
- Polineuropatia: Motosensoria (20%)
- Cute: Decolorazione scura delle estremità
- Decorso
- Variabilmente progressivo
- Sopravvivenza media: 6 ÷10 anni
- Laboratorio
- Disfunzioni della vescica (> 90%)
- EMG: Denervazione (20%)
- Patologia
- Perdita neuronale e gliosi
- Extrapiramidale: Putamen; Sostanza nera; Locus ceruleus
- Altre tronco cerebrale: Oliva inferiore; Pontine nuclei
- Cellule di Purkinie
- Midollo spinale: Cellule della colonna intermediolaterale (Toracica); Nucleo di Onuf
- Inclusioni citoplasmatiche gliali (argirofili): Sistemi motori soprasegmentali; Sistema autonomo sopraspinale
- Patologia dell'autonomo
- Midollo spinale: Ridotti i neuroni pregangliari ortosimpatici nelle
cellule della colonna intermediolaterale
- Midollo
- Ridotti i neuroni catecolaminergici (A1/C1) nella formazione reticolare intermedia ventrolaterale
- Normali proiezioni
- Discendenti: Cellule della colonna intermediolaterale nel midollo spinale
- Ascendenti: Vasopressina neuronale nell'ipotalamo
- Collegamento esterno:
eNeurology
Riley-Day, ovvero giorni storti (HSAN 3) 19
l
Inibitore
del gene del polipeptide leggero κ potenziatore nelle cellule B, proteina
chinasi associata ai complessi (IKBKAP)
; Cromosoma 9q31;
Recessiva
- Mutazioni IKBKAP
- Transizione da T a C nella coppia di basi 6 nel sito donatore di splicing dell'introne
20 (2507+6T-C)
- Aplotipo maggiore: Presente nel 99,5% degli alleli della malattia
- Aplotipo ancestrale comune: Effetto fondatore
- Frequenza dei portatori nella popolazione ebrea askenazita: 1/36
- Effetto della mutazione
- Lacune variabili nell'esone 20
- Messaggio di tipo selvatico espresso in modo tessuto-specifico
- Linfociti: Proteina di tipo prevalentemente selvatico
- Cervello: Principalmente proteina mutante
- Arg696Pro: Mutazione missenso
- Rara
- Tutti i pazienti eterozigotici per l'aplotipo maggiore
- La mutazione preannuncia la distruzione del sito della potenziale
fosforilazione della treonina
- Associato con fenotipo lieve
- Pro914Leu14
- Trovata in un paziente non ebreo (....o corna
....o non cercata, ..lapidazione ..o screening? ndt)
- Sindrome altrimenti clinicamente tipica
- La mutazione distrugge la fosforilazione della proteina
- Proteina IKBKAP
- Membro del complesso contenente altre proteine non identificate
- Localizzazione: I livelli più elevati della proteina IKBKAP nel cerebello, talamo, pituitaria, testicoli
- Funzione: ? Ruolo nei meccanismi di attivazione dei geni
- Regolazione e attivazione della risposta allo stress attraverso i
percorsi di segnalazione c-Jun terminale N delle
chinasi (JNK)
- Mutante: Può alterare l'interazione della IKAP con JNK
- Errata regolazione di JNK
- Sviluppo e differenziazione inadeguati
- Gene mutante espresso variabilmente
- Alla maggior parte delle degli mRNA nel cervello manca l'esone 20
- mRNA di tipo selvatico più abbondanti nei fibroblasti
- La completa assenza di espressione in tutti tessuti può essere letale
- Epidemiologia: La maggior parte dei pazienti sono ashkenaziti
- Caratteristiche cliniche
HSAN 3: Diagnosi clinica
| Fuoriuscita di lacrime emozionale |
Assente |
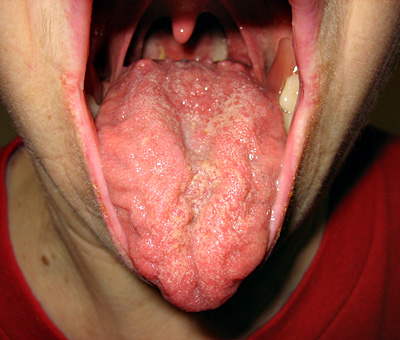
| Papille fungiformi linguali |
Assenti |
| Riflessi tendinei patellari |
Assenti |
| Arrossamento assonale dopo istamina ipodermica |
Assente |
|
Ereditarietà negli askenaziti |
Presente |
|
- Insorgenza: Congenita
- All'inizio del decorso della malattia
- Neonati: Ipotermia; Crisi di vomito; Ipotonia
- Occhi
- Flusso lacrimale (Fuoriuscita di lacrime): Ridotta o assente (99%)
- Iperestesia ed erosioni cornee
- Papille fungiformi linguali: Assenti
- Sensazioni
- Dolore Insensibilità
- Temperatura: Soglie anormali per il caldo e per il freddo
- Modalità a fibre grosse relativamente rispBracciaiate
- Distribuzione: Tronco ed estremità inferiori maggiormente colpiti
- Sensazioni viscerali preservate
- Riflessi tendinei profondi: Assenti
- Crisi dell'autonomo
- Generale: Irritabilità, febbre, svenimenti
- Ipotensione posturale: Nessuna tachicardia compensatoria
- Crisi
- Ipertensione
- Tachicardia
- Iperidrosi
- Alterazioni comportamentali
- Trattamenti: Diazepam (Vomito); Clonidina (Ipertensione)
- Cute chiazzata (99%)
- Sudorazione: Aumentata (99%)
- Normali le risposte cutanee ortosimpatiche
- Disturbi gastrointestinali
- Disfunzione gastrointestinale: Dismotilità
- Esofagea e gastrica
- Disfagia
- Riflusso gastrofaringeo
- Crisi di vomito
- Scheletro
- Scoliosi: Giovanile; Curvatura a sinistra più comune che nella scoliosi idiopatica
- Traumi: Fratture e necrosi asettica
- Respiratorio
- Anormali risposte agli stati ipossici e ipercapnici
- Aspirazione: Polmoniti ricorrenti
- Malattia polmonare restrittiva
- Sonno: Apnea e ipopnea centrali
- Respiro trattenuto
- Cardiovascolare: Ipotensione posturale
- Renale: Ischemia; Azotemia pre-renale
- Pazienti più anziani
- Distrofia delle unghie delle dita
- Gusto: Ridotto, specialmente il dolce
- Perdita della vibrazione: Con l'aumentare dell'età (> 13 anni)
- Atassia: Disturbi dell'andatura
- Sindromi psichiatriche
- Decorso
- Solitamente fatale: Morte nel 50% < 30 ÷ 40 anni
- Cause della morte: Polmonare; Renale; Morte improvvisa
- Altro
- Sindrome con caratteristiche specifiche della RD: Mancanza della fuoriuscita lacrimale;
Insorgenza precoce della ipotensione posturale
- Altri disturbi degli askenaziti: Fibrosi cistica, Tay-Sachs, malattia di Canavan
- Laboratorio
- Iponatremia: Durante le crisi
- Istamina intradermica: Arrossamento assente
- Catecolamine
- Rapporto HVA/VMA: Alto
- Rapporto DOPA/DHPG: Alto nel plasma
- Rimanendo in piedi: Bassi aumenti dei livelli plasmatici di NE e dopamine
idrossilasi b
- Crisi emotive: Livelli plasmatici molto alti di NE e DA
- Patologia
- Assoni non mielinizzati
- Marcata riduzione progressiva: 5% ÷ 15% del normale
- Ridotto numero dei corpi cellulari nei gangli della radice dorsale
- Perdita di neuroni dell'autonomo
- Gangli ortosimpatici cervicali superiori piccoli
- Gangli parasimpatici: Generalmente normali; Ad eccezione dei gangli sfenopalatini
- Relativamente rispBracciaiati i grossi assoni: 65%
÷ 100%
- Midollo spinale
- Ridotta principalmente la sostanza P assonale nella sostanza gelatinosa
- Colonne grigie intermediolaterali: Neuroni ridotti
Neuropatia sensoria congenita con anidrosi (HSAN 4)
l
Recettore TRKA/ NGF (NTRK1)
; Cromosoma 1q21-q22;
Recessiva
- Nosologia: Insensibilità congenita al dolore con anidrosi (CIPA)
- Mutazioni genetiche
- Tipi: delezioni, sito di splice, frame shift, e missenso
- Identificate > 40: La maggior parte delle sono uniche
- Domini: Peptidi segnale, extracellulare e intracellulare
- Alta prevalenza della malattia nei beduini arabi: Dovuta alla mutazione
1926-ins-T
- Mutazioni nello stesso gene associate con: Carcinoma tiroideo midollare
famigliare
- Conseguenze funzionali delle mutazioni
- Dominio extracellulare: Può impedire il legame del fattore di crescita
nervosa con il recettore
TRKA
- Dominio intracellulare: Può interferire con la trasduzione del segnale
- Proteina
- Domini singolo extracellulare, transmembrana e intracellulare
- Dominio extracellulare: Legante a NGF; Peptidi segnale
- Dominio intracellulare: Tirosina chinasi
- Caratteristiche cliniche: Omogenea
- Insorgenza
- Congenita: Ipotonia
- Fanciullezza: Pietre miliare motorie ritardate; Ipoidrosi; Fratture
senza dolore
- Insensibilità congenita al dolore
- Occhi
- Diminuzione della sensibilità e dei riflessi corneali
- Lacrimazione emozionale: Presente
- Sensazioni: Ridotti dolore, temperatura e viscerali
- La sensazione dolorosa rispBracciaiata parzialmente in alcune regioni del
- Ponte nasale
- Orecchie: Dietro le orecchie; Meato uditivo esterno
- Area cutanea cervicale posteriore: Alcuni pazienti
- Sensazione della temperatura: Assente o ridotta
- Riflessi tendinei: Normali, aumentati o diminuiti
- Autonomo
- Assenza di sudorazione
- Cute chiazzata
- Normale: Controllo della pressione sanguigna; Motilità gastrointestinale; Fuoriuscita lacrimale
- CNS
- Automutilazione
- Deformità delle articolazioni: Ginocchia e caviglie
- Bocca: Punta della lingua; Labbra
- Cute: Con cicatrici e ustioni
- Sistemico
- Febbre
- Idiopatica
- Episodica e ricorrente
- Malattia a manifestazione precoce: 3 mesi
- Iperpnea episodica
- Distrofia delle unghie delle dita
- Guarigione delle ferite: Lenta
- Decorso
- Morte precoce per iperpiressia: Fino a 20%
- Setticemia
- Laboratorio
- Esami provocativi 8
- Istamina: Ponfi evocati; Nessuna risposta di arrossamento assonale (100%)
- Mecolile: Nessuna lacrimazione
- Pilocarpina: Pilocarpina
- Risposte cutanee ortosimpatiche: Assenti (100%)
- Velocità di conduzione nervosa: Normale
- Anemia (79%)
- Patologia
- Nervi periferici
- Assoni sottili mielinizzati e non mielinizzati: Assenti; 0%
÷ 5% del normale
- Assoni grossi mielinizzati: Lievemente ridotti; 45%
÷ 65% del normale
- Gangli delle radici dorsali
- Neuroni sensori sottili assenti: Associato con l'insensibilità al dolore
- Ghiandole sudoripare: Assenza di innervazione delle ghiandole escretrici
- Ipotrofiche o assenti nel derma
- Associato con anidrosi e ipertermia
- Cute: Ridotta innervazione; Assoni rimanenti colorazione per GAP-43
Malattia di Parkinson ad insorgenza giovanile
l
Parkina
; Cromosoma 6q25.2-q27;
Recessiva
- Genetica: Mutazioni
- Localizzazione: Esoni 2 ÷ 9; Più frequenti nell'esone
7
- Delezioni piccole e missenso
- Pazienti spesso con composizione eterozigotica
- Insorgenza: Numerose sindromi 17
- Età: 7 ÷ 64 anni; Solitamente < 40 anni; Più tardi in 1 paziente con
genitori consanguinei
- Sindromi all'insorgenza
- Sindrome di Parkinson: Tremore
- Distonia: Indotta dall'esercizio o cervicale
- Periferico
- Disfunzione dell'autonomo: Può essere presente da sola nel 60%
- Neuropatia periferica: assonale
- Clinica
- Parkinson: Bradicinesia; Rigidità; Acinesia
- Tremore: Gambe e mani
- Distonia: Specialmente nei piedi
- Discinesie: Con trattamento; 100%
- Riflessi tendinei: Solitamente normali; Occasionalmente vivaci
- Autonomo (60%): Stimoli 45%; Impotenza 28% dei maschi; Svenimenti ortostatici 13%
- Corticale
- Psicosi
- Funzioni cognitive: Normali
- Decorso: Lenta progressione
- Risposte al trattamento: L-DOPA; Anticolinergici
l
Cromosoma 9q34; Recessiva
- Caratteristiche cliniche
- Neonatale
- Ptosi e apertura degli occhi ritardata
- Ipotensione, ipoglicemia, e ipotermia
- Infanzia e adulti
- Progressive ipotensione posturale con esercizio
- Intasamento nasale
- Eiaculazione prolungata o retrograda (erezione normale)
- Tolleranza all'esercizio ridotta
- Ptosi
- Laboratorio
- Carenza di dopamina idrossilasi beta
- Noradrenalina e adrenalina basse in plasma, urine, e CSF
- Aumento della dopamina in plasma, urine, e CSF
- Trattamento: L-dopa - Elevati livelli di noradrenalina
l
Cromosoma 11p15.5; Recessiva
- Caratteristiche cliniche in 1 paziente
- Iperattività dell'autonomo
- Dermatografismo
- Eccessiva sudorazione
- Ridotta temperatura corporea
- Tachicardia sinusale intermittente
- Neuromi acustici
- Obesità
- Personalità caratterizzata dalla ricerca di novità
Ipotensione ortostatica ereditaria
l
Cromosoma 18q; Dominante
- Nosologia: Iperbradicinismo; Streeten tipo
- Genetica: Pazienti occasionali non penetranti
- Insorgenza: Leggere emicranie nella posizione eretta
- Clinica
- Colorazione delle caviglie: Blu porpora
- Pressione sanguigna
- Sistolica: Ridotta; Ortostatica cala nella BP sistolica (>18 mm/Hg)
- Diastolica: Aumentata
- Tachicardia
- I segni ed i sintomi si risolvono quando il paziente è supino
- Progredisce a sincope e palpitazioni
- Trattamenti: Propranololo, fluorocortisone o ciproeptadina
- Laboratorio
Aniridia di tipo 2
l
Geni box appaiati 6 (PAX6)
; Cromosoma 11p13;
Dominante
- Pax6
- Famiglia dei geni box appaiati
- Regolatore trascrizionale coinvolto nella oculogenesi
- Governa la proliferazione cellulare e la migrazione dei neuroni 'nati successivamente'
- Altri tessuti: Pancreas; Pituitaria anteriore
- Clinica
- Occhi
- Cataratta
- Nistagmo
- Acuità visiva: Ridotta; Ipoplasia della fovea; Glaucoma
- Controllo della pressione sanguigna: Anormale
- Disturbi della termoregolazione
- Olfatto: Ridotto
- Altre sindromi PAX6
- Anomalia di Peters
- Ectopia delle pupille
- Ipoplasia della fovea
- Ipoplasia o aplasia dei nervi ottici
- Cheratite
- PAX6: 2 mutazioni
- Insorgenza: Infanzia
- Craniofacciale: Non occhi; Capo piccolo; Orecchie grandi; Ponte nasale appiattito,
atresia delle coane e narici piccolissime
- CNS: Assenti ottica nervi e chiasma; cervello piccolo; Assenza del corpo calloso
- Morte: Prima di 1 mese di vita
- Altre aniridie: Sindrome di Gillespie
- Modello animale: Topo dagli occhi piccoli (Sey)
Sindrome acalasia-addisonianismo-alacrimia (AAA) (Allgrove)
11
l
AAAS (Aladina)
; Cromosoma 12q13;
Recessiva
- Epidemiologia
- 7 famiglie: Nordamericane, nordafricane e europee
- Storia familiare: Famiglie consanguinee o sporadica
- Mutazioni del gene AAAS
- Localizzazione: Tutte le parti del gene
- Troncamento e missenso
- Composizione eterozigotica
- Una mutazione è una frameshift
- Seconda mutazione
- Missenso: Malattia più lieve
- Stop o mutazione in regioni importanti della proteina: Malattia più grave
- Associata con famiglie aventi pieno fenotipo AAA
- Sindrome AAA avente eterogeneità genetica
- Proteina aladina
- Tipo: Famiglia di ripetizioni WD delle proteine di regolazione
- Espressione genetica
- Cerebrale: ? Ruolo nello sviluppo dei PNS e CNS
- Neuroendocrine: Ghiandole adrenali
- Tratto gastrointestinale
- Localizzazione subcellulare:
Nucleare
- Diretta ai complessi poro nucleari (NPC; Siti del trasporto nucleocitoplasmico)
- Proteina mutata
- Comune: Non si riesce a localizzare gli NPC
- Altre: ? Anormalità delle funzioni dei pori nucleari
- Clinica
Di M Al-Lozi
|
- Insorgenza: Solitamente infanzia
- GI
- Acalasia; Atonia gastrica
- Difficoltà ad alimentarsi
- Xerostomia; Chelite angolare
- Lingua: Glossite; Fessurata
- Endocrino
- Cute
- Iperpigmentazione
- Eccessive pieghe nei palmi delle mani e piante dei piedi
- Ipercheratosi
- Faccia: Lunga magra; Philtrum lungo; Labbro superiore stretto
- Scheletro: Bassa statura; Osteoporosi; Microcefalia
- Occhi
- Alacrimia: La più coerente caratteristica clinica
- Atrofia ottica
- Disturbi pupillari: Pupillotonia
- Neuropatia periferica: Motorio-sensoria o motoria; Perdita assonale
- CNS
- Disfunzione dell'autonomo
- Parasimpatico e ortosimpatico
- Cardiovascolare
- Sudomotoria
- Sindrome variante: Insorgenza adulta, disturbo simile alla SLA
27
- Età di insorgenza: Adolescenza o adulta
- Bulbare
- Disartria
- Disfagia
- Lingua spastica
- Debolezza
- Insorgenza: Gambe
- Prossimale e distale
- Simmetrica
- Neuropatia motoria
- Distribuzione: Gambe; Braccia (ulnare)
- Atrofia: Ipotenare e muscoli altra mano; Parto
- Fascicolazioni: Arti e lingua
- Motoneuroni superiori
- Spasticità: Tutti gli arti
- Riflessi tendinei: Vivaci, eccetto che alle caviglie
- Autonomo
- Acalasia
- Disautonomia
- Alacrimia
- Ipotensione ortostatica
- Impotenza
- Iperidrosi
- Endocrino: Insufficienza adrenale,
ad insorgenza successiva
- Altre
- Lingua plicata
- Ipercheratosi plantare
- Alacrimia
- Faccia: Radici nasali ipertrofiche; Ciuffetto anteriore
- Progressione: Lenta
- Laboratorio
- NCV
- Ampiezza di CMAP: Piccola
- Velocità normale
- SNAP: Normale
- EMG: Denervazione cronica
- MRI del cervello: Atrofia cerebrale , moderate
- Laboratorio
- Episodi di ipoglicemia
- NCV
- Predominantemente coinvolgimento motorio
- Perdita assonale (CMAP piccoli); Lieve rallentamento
- Surale: SNAP ridotti con l'età
- ACTH: Alta
- Patologia
- Adrenale: Zona fascicolata adrenale assente; Zona glomerulosa quasi normale
- Esofago inferiore: Assenza delle cellule gangliali e degli assoni
- Morfologia nucleare: Normale
Distrofia autoimmunitaria poliendocrinopatia-candidiasi-ectodermica (APECED)
l
Regolatore autoimmunitario (AIRE); Cromosoma 21q22.3; Recessiva
- Insorgenza: 1a decade
- Clinica
- Endocrino:
- 1° Ipoparatiroidismo
- 1° Ipogonadismo
- Insufficienza adrenale
- Diabete mellito: Insulino-dipendente
- Ipotiroidismo: Latente
- Ipoaldosteronismo: Transitorio, isolato
- Insufficienza gonadica: Insufficienza primaria delle cellule di sertoli
- Capelli: Alopecia totale
- Denti: Ipoplasia dello smalto dentale
- Occhi: Cheratopatia; Cheratocongiuntivite
- Gastrointestinale: Malassorbimento; Diarrea; Epatite cronica attiva; Acalasia
- Moniliasi
- Laboratorio
- Anemia perniciosa: Ad insorgenza giovanile
- Immunità umorale: Ipogammaglobulinemia; antiadrenale, antitiroidea e
anticorpi antinucleari
- Immunità cellulare: Basso rapporto cellulare T4/T8
Sindrome della tachicardia posturale ortostatica (POTS)2
- Definizione
- Intolleranza ortostatica sintomatica
- Associata con incremento del ritmo cardiaco di almeno 30 battiti/minuto
- Pazienti caratteristici
- Giovani: Media 29 anni; Gamma 15 ÷ 50 anni
- Femmine:maschi: 4:1
- Insorgenza
- Decorso: Acuta o subacuta
- Prodromo: 50% dei pazienti hanno sintomi di un antecedente malessere virale
- Sintomi
- Compaiono con la posizione eretta
- Si risolvono con positura supina
- 3 o più sintomi presenti per > 3 mesi
- Debolezza: Estremità inferiori o diffusa
- Vertigini o bagliori
- Visione sfocata
- Palpitazioni
- Difficoltà respiratorie
- Nausea
- Cefalea
- Altri sintomi
- Prodromo: Infezione (50%)
- Intolleranza all'esercizio: Affaticamento
- Sincope
- Occhi o bocca secchi
- Gastrointestinale: Gonfiore; Sazietà precoce; Diarrea e costipazione
- Sintomi aggravati da: Calore; Pasti; Esercizio
- Segni
- Ritmo cardiaco: Aumenta d > 30 bpm nella posizione eretta
- Ristagno venoso nelle gambe nella posizione eretta
- Sudorazione nelle gambe: Ridotta
- Laboratorio: Parziale denervazione dell'ortosimpatico, specialmente nelle gambe
5
- Esaminazione inclinata: Eccessivo incremento del ritmo cardiaco
- 120 ÷ 170 bpm all'inclinazione verso l'alto
- Livello raggiunto dopo 2 minuti
- Esame adrenergico
- Stimoli (Pressorio freddo; Na nitroprussiato; Tiramina): Ridotto aumento
del livello della norepinefrina nelle gambe
- Livelli plasmatici della norepinefrina
- Supino: Normale o basso (60% del normale)
- In posizione eretta: Aumenta a > 600 pg/ml
- Manovra di Valsalva: Scompensata la funzione vasocostrittrice periferica baroriflessa mediata
-
Esami cardiovagali: Normali
- Sudorazione: Ipoidrosi distale, postgangliare
- Storia familiare di intolleranza ortostatica
: 27%
l
SLC6A2 (Trasportatore della norepinefrina trasportatore)
; Cromosoma 16q12.2;
Dominante
- Mutazione: Missenso Ala457Pro
- Proteina trasportatore della norepinefrina mutata: Non funzionale
- Caratteristiche cliniche
- Alterata regolazione del ritmo cardiaco
- Laboratorio: Metabolismo della norepinefrina alterato
- Penetranza variabile
- Trattamento
- Espansione del volume
- Dieta: Ad alto contenuto salino
- Elevata assunzione di liquidi: Bere rapidamente acqua, 120
÷ 480 ml in 5 minuti
- Agonisti degli adrenergici β: Basso dosaggio
- Denervazione dell'ortosimpatico: Midodrina, basso dosaggio
- Contromanovre fisiche
- Prognosi
- Miglioramento nella maggioranza dei casi (80%): 60% possono diventare
normali
- Migliore con storia di infezioni prodrome
Ipotensione in nell'anziano 6
- Fattori associati
- In posizione eretta: L'ipotensione aumenta con la durata della
posizione eretta
- Post-prandiale
- Alto contenuto di carboidrati nei pasti
- Temperature elevate
- Farmaci antiipertensivi
- Clinica: Può essere associata con sincope
Leucodistrofia ad insorgenza in età adulta (ADLD)3
l
Lamina B1
; Cromosoma 5q31;
Dominante
- Epidemiologia: Famiglie irlando-americane
- Genetica 25
- Mutazione: Duplicazione del gene Lamina B1
- Proteina Lamina B1
- Ampiamente espressa
- Componente della lamina nucleare
(involucro)
- Mutazione: Sovra-espressione nel cervello
- La sovra-espressione causa una anormale morfologia nucleare: Bolle e
ripiegamenti della membrana
- Insorgenza
- Adulti: 30' ÷ 50'; Media 41 anni
- Prima caratteristica dell'autonomo
- Clinica
- Autonomo
- Disfunzioni intestino/vescica
- Impotenza (nei maschi)
- Ipotensione ortostatica
- Ipoidrosi
- Piramidale
- Perdita delle abilità motorie fini
- Paraparesi spastica ~90%
- Disfunzione della colonna posteriore del midollo spinale
- Cerebellare
- Nistagmo 69%
- Atassia 100%
- Decorso
- Lentamente progressivo in 20 anni
- Perdita dei movimenti volontari
- Fatale
- Diagnosi differenziale
- Sclerosi multipla progressiva
- Demielinizzazione tossica: Esaclorofene; Cuprizone
- MRI del CNS: Perdita di mielina diffusa simmetrica
- Iniziante nei lobi frontali del cervello
- Si estende al cervelletto
- Neuropatologia
- Localizzazioni: Materia bianca del centro semiovale e dei peduncoli cerebellari
- Perdita di mielina in chiazze isolate e confluenti
- Microscopia
- Vacuolazioni della materia bianca
- Oligodendrociti abbondanti dentro le lesioni
- Astrociti sparsi: Con processi di anormale accorciamento e imperlinatura
- Nessuna infiammazione o chiara patologia neuronale
Sindrome della sudorazione indotta dal freddo
12
l
Fattore simile al recettore delle citochine 1 (CRLF1)
; Cromosoma 19p12; Recessiva
- Mutazioni genetiche
- Più grave famiglia norvegese: 844_845delGT
- Famiglia israeliana: 2 mutazioni missenso; R81H e L374R
- Avviene anche nella: Sindrome di Crisponi
- Proteina CRLF1
- Espressa nei muscoli scheletrici e altri tessuti
- Recettore delle citochine solubili: Omologo ai recettori di tipo 1
- Associata alle citochine simili alle cardiotrofine
- Compete con CNTF per il legame al recettore CNTF
- Sindromi coinvolgenti proteine correlate
- Epidemiologia: Famiglie israeliane e norvegesi
- Insorgenza: Variabile
- Famiglia norvegese: Neonatale ; Disturbi di alimentazione e respiratori
- Clinica
- Sudorazione
- Insorgenza: 1a o 2a decade
- Indotta da temperature 7°C ÷ 18°C
- Distribuzione anatomica
- La sudorazione inizia in regioni localizzate: Presternale; Mani
- Diventa profusa su grandi segmenti: Sopra la cintola; A chiazze sulla schiena e sulla gabbia toracica
- Pilocarpina nelle aree affette con caldo o febbre
- Decorso: Non progressivo
- Perdita sensoria: Modalità a fibre sottili
- Nei pazienti colpiti più gravemente
- Ridotto il dolore indotto da calore e interventi chirurgici
- Ridotta sensazione di caldo in ambienti caldi
- Alimentazione: Ridotto interesse al cibo
- Scheletro
- Palato ad arco alto: Voce nasale
- Faccia
- Ridotte espressioni facciali
- Ponte nasale depresso
- Apertura della bocca limitata
- Contratture del gomiti
- Clinodattilia
- Cifoscoliosi: Toracolombare
Sindrome della sudorazione indotta da freddo 2
l
Simili alle cardiotrofine citochine fattore 1 (CLCF1)
; Cromosoma 11q13.3;
Recessiva
- Epidemiologia: Singolo paziente autraliano
- Genetica: Mutazioni eterozigotiche; Tyr107Ter e Arg197Leu
- Proteina CLCF1
- Famiglia: Citochine interleuchina 6
- Sindromi coinvolgenti le proteine correlate
- Clinica
- Insorgenza: Infanzia; Difficoltà ad alimentarsi; Sudorazione
- Sudorazione
- Indotta dal freddo
- Incapacità a sudare con il caldo
- Localizzazione: Specialmente faccia, tronco e arti superiori
- Faccia
- Debolezza: Lieve
- Orecchie: Situate ad angolo retto dal cranio
- Polineuropatia
- Predominantemente sensoria
- Perdita assonale
- Scheletrico e morfologico
- Palato: Palato ad arco alto
- Gomiti: Deformità valga; Incapacità ad estendere completamente
- Clinodattilia: Dita delle mani e dei piedi, sindattilia del seconda e terzo dito di entrambi
i piedi
- Scoliosi toracolombare
- Lordosi lombare
- Malattia degenerativa della colonna vertebrale cervicale e lombare
- Decorso: Non progressivo
Sindrome arlecchino
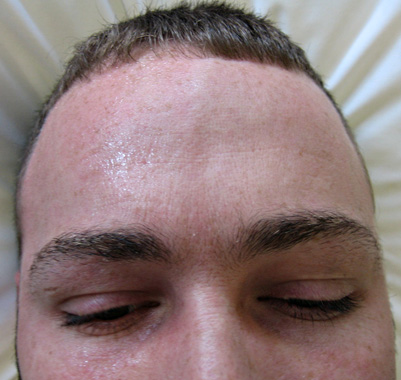
Dalla: M Al-Lozi
|
- Clinica
- Ipoidrosi: unilaterale
- Iperidrosi: Lato normale
- Arrossamento: Lato normale
- Indotta da: Calore o Esercizio
- Insorgenza: Può essere improvvisa
- Decorso: Benigno
- Occasionali
- Braccia coinvolgimento, omolaterale o controlaterale
- Può sovrapporsi a: Sindrome di Ross;
Sindrome di Horner
- Sindrome parasomina sovrapposta
Cause
- Idiopatica
- Infarto del tronco cerebrale
- Neurinoma: Mediastinale superiore
- Cateterizzazione della vena giugulare interna
Patologia possibile
- Assoni ortosimpatici cervicali pre- o post-gangliari
- Neuroni parasimpatici: Gangli ciliari
Malattia con inclusioni neuronali intranucleari (ialine) (NIID)
l
Dominante o sporadica
- Insorgenza
- Età: Solitamente infanzia
- Disturbi dell'autonomo
- Clinica
- Insufficienza dell'autonomo
- Incontinenza: Fecale; Urinaria
- Posturale: Ipotensione; Bagliori; Sincope
- Disfunzione erettile
- Ipoidrosi
- GI: Pseudoostruzione
- Decorso: Può essere progressiva o episodica
- Disfunzione cerebellare
- Atassia: Andatura
- Disartria
- Dismetria oculare
- Neuropatia periferica: Alcuni famiglie
23
- Motorio: Debolezza della faccia e degli arti; Voce nasale; Disfagia
- Perdita sensoria: Panmodale; Distale
- Elettrofisiologia: Perdita assonale
- Patologia nervosa
- Perdita assonale: Sottili > grossi
- Inclusioni nelle cellule di Schwann: Colorazione con ubiquitina
- Motorio
- Debolezza: Gambe o diffusa
- Ipotonia
- Riflessi tendinei: Aumentati o ridotti (con neuropatia)
- Laboratorio
- Inclusioni patologiche (Biopsia rettale): Eosinofile, ubiquitinate,
nucleari neuronali
- MRI: Atrofia cerebellare ± corticale
- EMG: Denervazione distale; Anormalità sfinteriche
- Biopsia muscolare: Normale
- CK serica: Elevata in alcuni pazienti
- Patologia
- Inclusioni nucleari neuronali e gliali: Generalizzate in CNS e PNS
- Perdita neuronale e gliosi: Neuroni dei gangli dell'ortosimpatico e sensori; Neuroni
motori spinali e tronco cerebrali
Demenza con corpi di Lewy 20
- Età di insorgenza: 65 anni
- Clinica
- Declino cognitivo: Progressivo
- Cognizioni fluttuanti o risvegliate
- Ricorrente formazioni di allucinazioni visive
- Parkinsonismo: spontaneo
- Disfunzione dell'autonomo
- Anidrosi: Distale
- Ipotensione, ortostatica
- Sintomi urinari (35%)

Di Horsley Gantt
|
Ritorna a
Neuromuscolare home page
Ritorna a
Indice delle polineuropatie
Ritorna a
Autonomo physiology ↦ FBracciaacologia
Riferimenti
1. Neuropediatrics 1996;27:26-31
2. Mayo Clinic Proc 1999;74:1106-1110
3. Human Molecular Genetics 2000;9:787-793
4. NEJM 2000;343:847-855,
Arch Neurol 2009;66:735-741
5. NEJM 2000;343:1008-1014
6. Ann Int Med 2000;133:533-536
7. Muscle Nerve 2001;24:292-296
8. Pediatr Neurol 2001;25:397-400
9. Am J Med 2002;112:355-360
10. Nature Genetics 2002;May
11. Brain 2002;125:2681-2690
12. Am J Hum Genet 2003;72:Online January
13. Acta Neurol Scand 2003;107:72–73, Clinica Neurophysiology 2003;114:7–16
14. Am J Med Genet 2003;Online March
15. J Clin Invest 2003;111:907–913, J Clin Invest 2003;111:797-799
16. Muscle Nerve 2003; Online February 2003;
Neurology
2009;73:1501–1506
17. Brain 2003;126:1279-1292
18. Umani Mol Genet 2003; Online October
19. Muscle Nerve 2004;29:352-363
20. Neurology 2004;62:1804–1809
21. Am J Hum Genet 2005;72:March
22. Am J Hum Genet 2005;77:Online May
23.
Neurology 2005;65;1538-1543
24.
Arch Neurol 2006 63:513-518
25.
Nat Genet 2006 Sep 3
26. Am J Hum Genet 2007; Online Mar
27. Eur J Hum Genet 2008
Jul 16;
Amyotroph Lateral Scler
2008 Jul 10:1-3
17 dicembre 2009
Va a Acronimi e sigle
Va a Mitolario in
italiano
Va a Mitolario italiano -
inglese
Va a Mitolario inglese -
italiano