|
Introduzione e
definizione
I
mitocondri sono organelli molto complessi che si ritrovano praticamente
in tutte le cellule del corpo. Un grande grado di complessità è dovuto
al fatto che tra le oltre mille proteine localizzate nei mitocondri, 13
sono codificate dal DNA mitocondriale (mtDNA), mentre le rimanenti sono
codificate dai geni del nucleo della cellula (nei cromosomi) e importate
nei mitocondri. Prima di inoltrarci nella ereditarietà della malattia mitocondriale, voglio precisare cosa questo studioso definisce
come "malattia mitocondriale". In questo contesto, la malattia
mitocondriale si riferisce a qualsiasi malattia risultante da una
carenza di qualsiasi proteina localizzata nei mitocondri che é coinvolta
nel metabolismo energetico. Pertanto, le carenze della catena
respiratoria (trasporto di elettroni), sia causate da carenza di una o
più proteine codificate dai mitocondri o dal nucleo, sono malattie
mitocondriali. Dunque, secondo questa definizione, malattie della
ossidazione degli acidi grassi (beta), carenza del ciclo di Krebs e del complesso piruvato deidrogenasi
sono malattie mitocondriali. Sebbene le proteine coinvolte siano
codificate dal nucleo, esse sono localizzate nei mitocondri e sono
coinvolte nel metabolismo energetico. Sebbene geneticamente diverse,
tutte queste malattie mostrano similarità cliniche, in quanto da esse
deriva uno stato di deficit energetico.
Sfortunatamente, le
molteplici malattie classificate come malattie mitocondriali hanno
diversi tipi di ereditarietà. Infatti, è stato dimostrato che
quasi ogni "modello" di ereditarietà conosciuto si ritrova nelle
malattie mitocondriali. Comunque, la maggior parte delle malattie
mitocondriali conosciute sinora sono ereditarie sia in maniera
autosomica recessiva che materna. Il modello di ereditarietà è
importante, in quanto può essere di aiuto per rispondere alle seguenti
domande:
-
Altri
membri della famiglia, sia esistenti che non ancora nati, sono a
rischio di sviluppare una malattia mitocondriale?
-
Quale è il
rischio (in percentuale)?
-
Quando si
ammaleranno gli altri probabilmente?
-
Quando gli
altri si ammaleranno, la loro gravità sarà pari alla mia o a
quella di mio figlio?
O perfino peggio?
-
Quali tipi
di problemi o malattie potranno colpirli?
Come vedrete, non tutte queste domande possono avere risposta alla data
odierna in tutte le famiglie dove qualcuno è stato colpito da una
malattia mitocondriale. Il primo passo è determinare il modello di
ereditarietà presente nella vostra famiglia. Questo può essere fatto in due
modi basilari:
-
Basandosi
su una diagnosi confermata: ad esempio, se la persona è portatrice
della mutazione del mtDNA "MELAS"
A3243G, il vostro dottore potrà essere sicuro che il
modello ereditario nella vostra famiglia è quello di tipo materno.
Ciò perché, questa mutazione del mtDNA è ereditata in linea
materna, sia che la madre mostri o meno i sintomi. Le carenze
MCAD e SURF1
sono
sempre ad ereditarietà autosomica recessiva.
-
Basandosi
sulla genealogia: Sfortunatamente, la maggior parte delle volte il
difetto esatto non può essere trovato. Comunque, in molti casi
possiamo indicare con una certa approssimazione il modello di
ereditarietà sulla base di una attenta analisi della storia
familiare. Comunque, questa è la sola
possibilità se ci sono altri membri della famiglia che hanno
malattie che sono dovute probabilmente a deficit di energia.
Come capirete dai modelli di ereditarietà esposti più sotto, tutti i
modelli di ereditarietà coinvolgono proteine codificate dal codice
genetico dei geni nucleari eccetto uno. Questa eccezione è rappresentata
dall'ereditarietà materna che coinvolge i geni del mtDNA e le proteine
codificate da questi. Sebbene lunga e complessa, le informazioni
presentate qui sono effettivamente solo le basi, ed esistono importanti
eccezioni. Queste informazioni non devono essere considerate una
alternativa ad un consulto genetico individuale con un professionista
competente.
*Torna su*
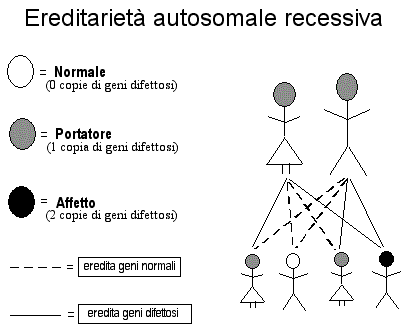
Ereditarietà autosomica recessiva
L'ereditarietà
autosomica recessiva potrebbe risultare familiare ad alcuni lettori in
quanto numerose malattie non mitocondriali sono ereditate in questo
modo, incluse la fibrosi cistica e l'albinismo. L'ereditarietà
autosomica recessiva è probabilmente il modello più comune nella
malattia mitocondriale, includendo disturbi della ossidazione degli
acidi grassi, del ciclo di Krebs, le subunità della
catena respiratoria codificate dal nucleo, ed i fattori di assemblaggio
e di trasporto. Ricordate che tutti noi abbiamo due copie di ogni gene
virtuale della codificazione nucleare, una da nostro padre e una da
nostra madre. Solo uno di questi geni a caso entra nel corredo che forma
l'uovo o lo spermatozoo. Nella ereditarietà autosomica recessiva, tutti
e due i genitori sono portatori in quanto essi hanno ambedue una copia
del gene che è difettosa. Essi non ne sono affetti perché hanno anche
una copia sana del medesimo gene. Se tutti e due, spermatozoo ed ovulo,
riportano il gene difettoso, alterato o mutante, il loro bambino non
avrà una coppia di geni che lavorano bene e normalmente, e manifesterà la
malattia.
Perciò, nella
ereditarietà autosomica recessiva, il 25% dei bambini erediterà il gene
difettoso da ambedue i genitori e manifesterà la malattia, il 50% dei
bambini erediterà un solo gene difettoso da uno dei genitori e diventerà
un portatore sano, come i genitori, ed il 25% dei bambini non erediterà
nessuna copia del gene difettoso e sarà quindi sano. Il
venticinque percento, o uno su quattro è il rischio per ogni bambino.
Pertanto in una scelta a caso, circa 1 su quattro dei fratellini o
sorelline potrà ereditare la malattia. La probabilità che qualcuno altro
al di fuori dei fratellini e sorelline, per esempio nipoti, cugini e
pronipoti erediti la malattia è molto piccola. Una eccezione importante
avviene in quelle famiglie dove i genitori sono consanguinei come
avviene nelle comunità isolate nelle quali la probabilità di
consanguineità è elevata.
Ci sono comunque delle eccezioni specialmente nei disturbi della
ossidazione degli acidi grassi, dove si possono avere comuni malattie ad
ereditarietà autosomica recessiva ad insorgenza infantile. Le malattie
che affliggono fratellini e sorelline sono normalmente simili per l'età
di insorgenza, gravità, ed i sistemi d'organi colpiti. Nel mondo, se un
successivo bambino eredita la malattia, lo farà molto probabilmente in
maniera molto simile, anche se non identica.
ESEMPIO:
In una famiglia con dieci figli, due
sorelle ed un fratello sviluppano attacchi epilettici gravi, ognuno a
pochi mesi di vita. Hanno gravi ritardi mentali, sebbene di diversa
gravità. Ambedue i genitori e gli altri membri della famiglia sono sani
senza problemi attribuibili a deficienza energetica.
*Torna su*
Ereditarietà materna
L'ereditarietà
autosomica recessiva era la cosa facile; l'ereditarietà materna,
conosciuta anche come ereditarietà mitocondriale o citoplasmatica, è la più complicata fra tutte. La
malattia mitocondriale ad ereditarietà materna non è rara, ed è
probabile che sia comune come malattia mitocondriale autosomica
recessiva. Tutte le malattie ad ereditarietà materna sono malattie
mitocondriali. Esempi includono la MELAS, la MERRF, la NARP e la LHON.
I figli
ereditano il loro DNA mitocondriale solo dalla loro madre, mentre il DNA
nucleare proviene dal padre e dalla madre. Le femmine trasmetteranno
sempre le loro mutazioni del DNA mitocondriale, errori o difetti
genetici, ed i maschi non trasmetteranno mai una mutazione del DNA
mitocondriale. Perciò, un bambino condivide le stesse sequenza del DNA
mitocondriale che posseggono i suoi fratelli e sorelle e la madre, ma
non con suo padre. In aggiunta, la madre, i fratelli e sorelle gli zii e
la nonna materna, e gli ascendenti in linea materna anche più lontani
condividono il medesimo DNA mitocondriale. In pratica, fratelli e
sorelle e madre sono spesso affetti da forme variabili di carenza
energetica, mentre gli ascendenti materni, zii e nonna ne sono affetti
solo talvolta.
A rendere la materia ancora più difficile c'è un concetto chiamato
"eteroplasmia". Mentre ognuna della nostre cellule contiene esattamente
2 copie di ogni gene nucleare, ogni cellula contiene un numero variabile
di copie del DNA mitocondriale, spesso molte migliaia per cellula. La
maggior parte delle persone "normali" hanno cellule omoplasmiche, ciò significa che le loro cellule contengono solo DNA
mitocondriale normale. Comunque, le persone con malattia mitocondriale
ad eredità materna ed i loro consanguinei materni hanno solitamente
cellule eteroplasmiche, significa che parte del DNA mitocondriale è
normale, ovvero non contiene mutazioni, e parte non è normale, contiene
mutazioni. Le proporzioni della eteroplasmia differiscono, spesso
drasticamente, all'interno dei consanguinei materni. Dunque, l'assunto
è che più è il DNA mutante in una cellula, più la cellula ha problemi; e
così è: in pratica la cellula funziona bene fino a che le proporzioni
del DNA mutante raggiungono un certo livello, dopo il quale non può più reggere,
e ne risulta la malattia. Questo livello varia ampiamente nei vari
tessuti, alcuni sono più sensibili alla carenza energetica, e per
differenti mutazioni.
Nell'insieme, questo
significa che i sintomi, la gravità, l'età di insorgenza, ecc.. di una
malattia mitocondriale possono variare tremendamente all'interno di una
famiglia. Così, sebbene una madre con mutazioni del DNA mitocondriale
passerà le mutazioni ai suoi figli, non tutti i bambini avranno
necessariamente i sintomi. In aggiunta, se i bambini hanno i sintomi, la
malattia che ogni figlio manifesterà potrà essere molto diversa,
dipendendo la gravità dalla percentuale de DNA mitocondriale mutante in
ogni parte del corpo. Questo crea un infinito numero di tipi di
manifestazioni della malattia mitocondriale. Per esempio, un ragazzo con
una grave malattia cardiaca può avere il 94% di DNA mitocondriale
mutante, contro il 6% normale nel cuore, ed il 34% nel cervello, mentre
sua sorella con epilessia può avere il 50% di mtDNA mutante nel cuore e
l'80% di mutante nel cervello.
Le
informazioni precedenti spiegano perché la malattia mitocondriale
ereditata maternamente, è molto variabile nei suoi effetti clinici.
Diversamente dalle malattie mitocondriali autosomiche recessive,
l'insorgenza della malattia mitocondriale ad eredità materna e
solitamente più tarda nella vita, includendo gli infanti, l'età
prescolare, l'età scolare, l'adolescenza, o l'età adulta. Può comunque
apparire molto simile alla malattia mitocondriale ad ereditarietà
autosomica recessiva, compresa grave malattia in infanti.
Non si possono
dare figurativamente accurate percentuali per la ricorrenza di questa
malattia in altri eventuali figli. Il rischio dipende dal livello
dell'ammontare delle mutazioni eteroplasmiche
nell'ovulo femminile, il quale è diverso fra ogni individuo ed è
praticamente impossibile da misurare. Spesso il rischio viene quotato
fra lo 0 ed il 100%, sebbene nell'esperienza di questo autore, molti
fratelli e sorelle siano affetti dal medesimo grado di gravità, mentre
una minoranza è affetta gravemente.
ESEMPIO: Un
ragazzo di 9 anni soffre di una forma intermittente di
disautonomia, attacchi epilettici, difficoltà
di apprendimento ed alla fatica. Sua sorella di 8 anni ha solo un grado
severo di svuotamento gastrico rallentato. Un fratello di 11 anni non è
malato. La loro madre ha epilessia a nuova insorgenza, emicrania, e
neuropatia periferica.
Ci sono
inoltre malattie mitocondriali conosciute come la LHON o neuropatia
ottica ereditaria di Leber nella quale la mutazione del DNA
mitocondriale che causa la mutazione, cecità acquisita, è omoplasmatica
- significa che tutti i mitocondri portano il difetto. Comunque, non
necessariamente una persona che ha una mutazione del DNA mitocondriale
LHON, sviluppa la cecità, lo fa solo il 10%. Confusi? Così é.
L'articolo seguente
della MDA spiega approfonditamente la genetica mitocondriale e può
essere letto in italiano all'indirizzo internet
http://www.fonama.org/i_mda.org/publications/quest/i_q64mito.html
o in
inglese all'indirizzo internet
http://www.mda.org/publications/quest/q64mito.html
*Torna su*
Ereditarietà recessiva collegata al cromosoma X
L'ereditarietà
recessiva può risultare familiare ad alcuni lettori in quanto molte
malattie molto ben pubblicizzate e che non sono malattie mitocondriali
sono ereditate in questo modo, incluse le forme più comuni di distrofia
muscolare, l'emofilia e il daltonismo (cecità ai colori). Una quantità
di malattie mitocondriali, le carenze dei più comuni tipi del complesso
della piruvato deidrogenasi (E1 alfa) sono collegate al cromosoma X.
Nella malattia
collegata al cromosoma X, il difetto del cromosoma X colpisce
solitamente solo i figli maschi. Questo poiché le femmine hanno 2
cromosomi X, 1 dalla madre ed uno dal padre, mentre i maschi hanno un
solo cromosoma X ereditato dalla madre, il cromosoma che fa il paio è il
cromosoma Y che ricevono dal padre. Le femmine con un cromosoma X sano
ed uno mutato non manifestano solitamente la malattia, per la presenza
di almeno un cromosoma sano. Comunque, queste femmine sono a rischio di
trasmettere una malattia genetica e sono chiamate "portatrici".
Dall'altro lato visto che i maschi hanno un solo cromosoma X, se questo
è mutato non dispongono della copia sana e svilupperanno la malattia
rappresentata dal gene difettoso
Se una femmina
portatrice ha figli, ci sono il 50% di probabilità che lei trasmetta un
gene difettoso ai suoi figli. Se sarà una figlia ad ereditare il gene
difettoso diventerà a sua volta portatrice. Se sarà un maschio ad
ereditare il gene difettoso, svilupperà la malattia.
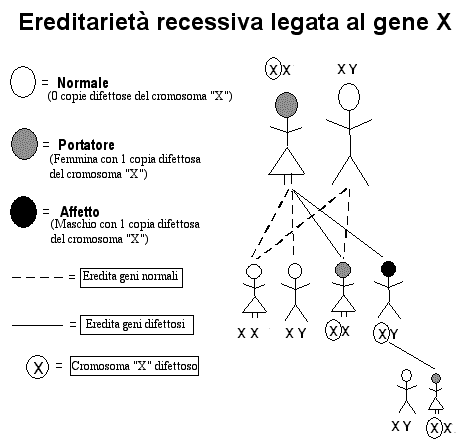
Si
ha una eccezione importante nel caso in cui alcune femmine portatrici
presentano, solitamente in forma lieve, la malattia. In casi rari la
malattia nei maschi è così grave da risultare letale ancora prima della
nascita, in tale caso si nota la malattia solo nelle femmine. Comunque,
in tutti i casi di malattia collegata al cromosoma X, ragazzi e ragazze
sono sempre colpiti in modo diverso, con i ragazzi colpiti in modo molto
più grave delle ragazze.
*Torna su*
Ereditarietà autosomica dominante
L'ereditarietà
autosomica dominante può essere familiare ad alcuni lettori come
caratteristica ereditaria delle malattie non mitocondriali come la
malattia di Huntington e l'ipercolesterolemia familiare. Tra le
malattie mitocondriali, una rara forma di sindrome di Kearn-Sayre
è ad ereditarietà autosomica dominante.
Nella
ereditarietà dominante, è sufficiente che solo un gene della coppia sia
difettoso perché la malattia si manifesti. Questo significa che ogni
persona con la malattia ha il 50% di probabilità di trasmettere la
malattia ai figli che potrà avere. Inoltre ogni figlio che eredita il
gene difettoso e che quindi può sviluppare la malattia, avrà a sua volta
il 50% di probabilità di trasmettere il gene difettoso. Comunque, con un
gene normale ed uno mutante, tutti questi individui possono sviluppare o
non sviluppare i sintomi della malattia. Se sviluppano la malattia,
questa potrà avere marcati gradi di gravità. Riguardo l'elevata
variabilità delle manifestazioni fra individui con un gene difettoso, le
ereditarietà autosomica dominante e la malattia mitocondriale ad
ereditarietà materna sono simili. Comunque, nella autosomica dominante i
ragazzi colpiti possono trasmettere la malattia ai loro figli, mai nella
ereditarietà materna.
*Torna su*
Casi sporadici — Dove non ci sono famigliari
colpiti
Nel
mondo reale, nella maggior parte dei casi, all'incirca nel 75% dei casi
il paziente è il solo membro della famiglia affetto da malattia
mitocondriale. Questi casi vengono chiamati
sporadici e presentano molte difficoltà nel rispondere alle domande che
riguardano l'ereditarietà.
La prima
questione è se il problema è dovuto alla genetica, alla gestazione, o ad
una combinazione dei due. Certamente gli aspetti genetici della malattia
mitocondriale sono ben conosciuti e sono brevemente riassunti sopra.
Comunque, non tutta la malattia mitocondriale e primariamente genetica.
Per esempio, i farmaci anti-retrovirali usati nella HIV o AIDS possono danneggiare i mitocondri e
causare i sintomi dovuti alla perdita di energia. Eliminando questi
farmaci il processo si inverte ed i sintomi si risolvono. Ci sono altre
cause ambientali della malattia mitocondriale, e molte di queste le
conosceremo più avanti.
Nella opinione di questo autore, la maggior parte
delle malattie mitocondriali sono probabilmente originate da ambedue le
cause: genetica E ambientale. Sempre nel caso dei farmaci anti-retrovirali, migliaia di individui non hanno problemi con l'uso di
questi farmaci mentre avviene per un pugno di loro. Ci sono
probabilmente ragioni genetiche per l'alta suscettibilità a questi
farmaci ed in alcuni casi infelici c'è una predisposizione genetica a
sviluppare la malattia. D'altro lato, nella MELAS, nella quale c'è una
chiara origine genetica primaria, il danneggiamento neurologico accade
spesso nel corso di malattie virali e o viene accelerato per lo scattare
di un grilletto ambientale su una malattia "genetica".
La seconda
questione è: se è genetica, la mutazione è stata ereditata? La risposta
è solitamente si, ma non sempre. Esistono mutazioni nuove o spontanee.
In particolare, La cancellazione, sezioni mancanti, nel DNA
mitocondriale tendono ad essere mutazioni nuove e non presenti nella
madre e nei fratelli. Comunque, le cancellazioni con duplicazione sono
spesso ereditate, ed alcune duplicazioni sono difficili da scoprire.
Tutto
questo significa che ci sono molte poche risposte nei molti casi in cui
una sola persona in una famiglia ha la malattia mitocondriale. La
condizione è probabilmente genetica, e può essere come non essere
ereditata. Possono essere coinvolti tutti e due i DNA, nucleare e
mitocondriale. L'ereditarietà è probabilmente autosomica recessiva,
materna, o sporadica ovvero non ereditata, ma non necessariamente.
Basandosi
su molte famiglie, alcuni gruppi danno una ricorrenza stimata del
rischio di malattia mitocondriale, La possibilità che ogni ulteriore
bambino dei medesimi due genitori sia colpito è del 10-15%. Questa è
probabilmente una stima grossolana, ma dovete discutere della
probabilità di ricorrenza per la vostra famiglia con un consigliere
genetico che abbia familiarità con la malattia mitocondriale.
- Written by Richard
Boles, M.D. e Terri Mason
*
Torna su *
|













